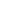
当敲除ETC复合物I关键亚基编码基因NDUFS2时,细胞对于GPX4抑制剂RSL3诱导的铁死亡表现出更高的敏感性,并且展现出更高水平的线粒体脂质过氧化。NDUFS2缺失增加了CoQ/CoQH2比例,并且在缺乏COQ2(CoQ生物合成关键酶)的细胞中,敲除NDUFS2对于细胞对铁死亡诱导剂的敏感性没有影响。作为NADH-醌氧化还原酶的酵母源NDI1,在将NADH2氧化为NAD+的同时,将CoQ还原为CoQH2,且在此过程中不进行质子泵送。在NDUFS2缺失细胞中,恢复表达NDI1可以消除由NDUFS2缺失引起的对RSL3增敏效应,这表明ETC复合物I抑制铁死亡的作用与其质子泵功能无关。因此,ETC复合物I的缺失通过减少线粒体中CoQH2的生成使细胞对铁死亡更为敏感,并且与其泵送质子或生成NAD+的功能无关。
有趣的是,ETC复合物I抑制剂不影响GPX4抑制诱导的铁死亡,这表明ETC复合物I抑制剂处理与NDUFS2缺失引起的铁死亡增敏效应存在差异。与NDUFS2缺失相比,ETC复合物I抑制剂处理不仅增加了CoQ/CoQH2比例,还导致ADP/ATP水平升高。产生这种差异的原因可能是NDUFS2的敲除过程中细胞伴随着生成ATP的补偿机制。ETC复合物I抑制剂处理导致的高ADP/ATP水平引发能量应激并诱导AMPK激活,抑制铁死亡的发生。而在AMPK缺失细胞中,ETC复合物I抑制剂处理显著增加了细胞对铁死亡诱导剂的敏感性。总的来说,ETC复合物I抑制剂处理与基因缺失细胞相比,AMPK激活的差异是导致不同铁死亡增敏效应的基础。
LKB1是AMPK的上游激酶,在多种肿瘤中发生突变,并且LKB1突变型肿瘤中呈现较低的AMPK活化水平。进一步研究表明,ETC复合物I抑制剂使五种不同LKB1-AMPK失活的移植瘤模型对铁死亡诱导治疗(放疗)更为敏感。这包括对照和AMPK抑制剂Compound C处理的HT-1080移植瘤模型;AMPK野生型和敲除的HT-1080移植瘤模型;LKB1野生型和敲除的NCI-H292移植瘤模型;LKB1野生型和突变型肺癌患者来源的移植瘤模型;以及Lkb1野生型和敲除的LKR-13同种移植瘤模型。以上结果表明,ETC复合物I抑制剂与放疗的组合可作为治疗LKB1突变型肿瘤的铁死亡诱导新策略(图1)。
参考文献
1. Stockwell, B.R.. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 185, 2401-2421 (2022).
2. Bersuker, K., Hendricks, J.M., Li, Z. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692 (2019).
3. Doll, S., Freitas, F.P., Shah, R. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698 (2019)
4. Mao, C., Liu, X., Zhang, Y. et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593, 586–590 (2021).
5. Wu, S., Mao, C., Kondiparthi, L., et al. A ferroptosis defense mechanism mediated by glycerol-3-phosphate dehydrogenase 2 in mitochondria. Proceedings of the National Academy of Sciences, 119, p.e2121987119 (2022).
6. Friedman, J.R., Nunnari, J.. Mitochondrial form and function. Nature, 505, 335-343 (2014).


公司主要产品有抗体制备全套产品、病毒包装 相关产品、2万多种科研用抗原抗体、近300 种病理级抗体、几千种重组蛋白,另有WB、 IHC、ELISA、细胞培养相关试剂及诸多特色 产品和特色技术服务。
 电话咨询
电话咨询
 在线咨询
在线咨询
 QQ
QQ
 二维码
二维码
 扫码二维码
扫码二维码